 FLICKR, HAFIZ ISSADEEN
FLICKR, HAFIZ ISSADEEN
Impervious pest
Species: House fly (Musca domestica)
Genome size: 691 million base pairs
The common house fly can be a major nuisance, but its abilities to tolerate pathogens and decompose waste make it a useful research subject. The house fly genome, published today (October 13) in Genome Biology, reveals expanded repertoires of genes involved in pathogen responses, detoxification, and chemosensing compared with the 123-million-base-pair genome of the fruit fly Drosophila melanogaster.
“House flies are a fascinating insect for scientists in many areas, such as developmental biology, sex determination, immunity, toxicology and physiology,” Jeff Scott, Cornell University entomologist and lead author of the paper announcing the sequencing feat, said in a press release. “The completed genome will be a phenomenal tool for researchers in all of these fields and will facilitate rapid advancements.”
M. domestica thrives on human and animal waste and is...
Flies are, however, hampered by a parasitoid wasp, Nasonia vitripennis, whose genome has also been sequenced. This host-parasitoid interaction could be a fruitful area for future investigations.
Pesticides have been used to keep house flies off farm animals for a century, and flies have repeatedly evolved resistance; insights into the genetic mechanisms of resistance could enable smarter approaches to pest control.
Little giant
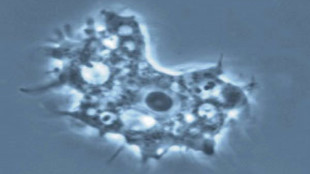 Microscope image of an Acanthamoeba cell.WIKIMEDIA, PESKYPLUMMER
Microscope image of an Acanthamoeba cell.WIKIMEDIA, PESKYPLUMMER
Species: Melbournevirus
Genome size: 369,000 base pairs
A virus isolated from an amoeba in a freshwater pond in Melbourne, Australia, is the sixth member of the “giant” virus family Marseilleviridae to be sequenced. Its genomic similarity to other Marseilleviridae allowed scientists at Aix-Marseille Université in France to evaluate selective pressure on the Melbournevirus’s 403 genes.
A high ratio of synonymous to non-synonymous mutations indicates that most of the genes are under purifying selection and thus are important for the fitness of the virus, the researchers reported earlier this month (October 1) in the Journal of Virology. “This conclusion contrasts with the more common view that many of the genes of the usually more diverse large DNA viruses might be (almost) dispensable,” the authors wrote.
The first known giant virus, Mimivirus, was found in the same type of amoeba, Acanthamoeba, in 2003. Mimivirus was so large (0.4 micrometers in diameter) that scientists first mistook it for a bacterium living inside the amoeba; its genome is 1.2 million base pairs long.
The study of giant viruses is reshaping scientists’ understanding of viruses, and suggests that amoebae, which eat bacteria and infect eukaryotic hosts, and are also infected by a variety of viruses and bacteria, may serve as “melting pots” for microbial evolution.
Matchless migrator
 WIKIMEDIA/USFWS, RICK HANSEN
WIKIMEDIA/USFWS, RICK HANSEN
Species: Monarch butterfly (Danaus plexippus)
Genome size: 273 million base pairs
An analysis of 92 monarch butterfly genomes from around the globe has upended assumptions about the species’ evolutionary origins, migration behavior, and pigmentation. The study, published this month (October 1) in Nature, suggests that monarchs originated in North America about 2 million years ago, and that populations in Central and South America, Europe, and the Pacific islands are far more recent.
Because most butterflies in the genus Danaus live in the tropics and do not migrate, scientists had presumed that North American monarchs split off long after the species first arose. However, a phylogeny of worldwide monarch populations, plus four other Danaus species, showed that monarchs likely originated in the southwestern U.S. or northern Mexico, and that their migrations are ancestral. Indeed, it appears that monarchs lost the migratory behavior three times in their evolutionary history.
This transformation was linked to changes in a single gene encoding a collagen protein important for flight muscle function. The researchers expected migrating monarchs to have stronger collagen. “They have to be big and tough and powerful,” study coauthor Marcus Kronforst, an evolutionary biologist at the University of Chicago, told the Los Angeles Times. “Then we did the experiments. And it turns out the migratory butterflies have less” collagen activity. “I thought: This doesn’t make any sense,” he said. “They should be doing more, not less.” Flight experiments demonstrated that the secret to successful migration lay in more efficient oxygen use.
The final surprise of the study came from examining the genome of a white monarch butterfly that lives in Hawaii. This revealed differences in a gene encoding myosin, which is known to transport pigment in vertebrate animals but was never before shown to play this role in invertebrates.
Expression exposer
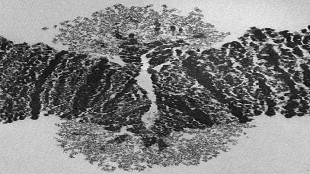 Transmission electron microscope image of chromatin in the BR2 “puff” of Chironomus tentans.BMC GENOMICS, ALEXEY KUTSENKO ET AL.
Transmission electron microscope image of chromatin in the BR2 “puff” of Chironomus tentans.BMC GENOMICS, ALEXEY KUTSENKO ET AL.
Species: Non-biting midge (Chironomus tentans)
Genome size: ~200 million base pairs
The first draft genome sequence of the tiny fly Chironomus tentans will aid studies of fundamental gene expression and chemical toxicity. The similarity of the C. tentans gene expression machinery to that of Drosophila melanogaster highlights the relevance of this midge as a model system, researchers from Stockholm University in Sweden reported last month (September 27) in BMC Genomics.
The larvae of C. tentans burrow into lake sediments, where they are often exposed to pollutants. In the laboratory, they are commonly used to test chemical toxicity. In addition, the C. tentans salivary glands contain unusual giant polytene chromosomes, which result from multiple rounds of DNA replication without cell division. The fly’s Balbiani ring (BR) genes are of particular interest because, during active transcription, they form a “puff” in the chromosome that allows the gene expression machinery to be readily studied in intact nuclei using in situ hybridization.
The genome sequence clarified the organization of the BR loci and identified a novel putative BR gene. This information enabled the researchers to visualize the organization of two of the BR genes in the BR2 puff, which revealed a unique architecture of unfolding chromatids during gene transcription.
Aquatic commodity
 FLICKR, ROBPOSSE
FLICKR, ROBPOSSE
Species: Common carp (Cyprinus carpio)
Genome size: 1.83 billion base pairs
The genome of the widely cultivated freshwater carp shows evidence of genome duplication and suggests genetic underpinnings for differences in the fish’s scales and skin color. The sequence will benefit breeding efforts for this relatively sustainable fish, which represents about one-tenth of global freshwater aquaculture production, a team of scientists from China, the U.S., and Hungary reported last month (September 21) in Nature Genetics.
By analyzing mutation rates within gene families, the researchers estimated that the carp lineage experienced a whole-genome duplication about 8.2 million years ago, resulting in 50 chromosomes, rather than the typical 24 or 25 found in most bony fishes.
After assembling a draft genome of the domesticated Songpu strain, the team resequenced 32 other carp specimens from both wild and domesticated populations in Asia, Europe, and the U.S. A phylogenetic analysis revealed distinct Asian and European clades, with US samples falling into both groups, reflecting multiple introductions of domesticated carp.
Transcriptome sequencing of skin from the Songpu and Hebao strains uncovered hundreds of differentially expressed genes. Two notable differences included a gene involved in hair follicle formation, reflecting a scale reduction in Songpu, and a gene required to transport cysteine into melanocytes. This gene showed higher expression in Hebao, which could explain its red pigmentation, the authors reported.
Interested in reading more?




