
 Left: Myxozoan spores (K. iwatai), each about 10 micrometres in diameter. Right: a moon jellyfish (Aurelia aurita), about 25 centimeters across.LEFT: UNIVERSITY OF KANSAS, A. DIAMANT; RIGHT: UNIVERSITY OF KANSAS, P. CARTWRIGHT (VIA EUREKALERT)
Left: Myxozoan spores (K. iwatai), each about 10 micrometres in diameter. Right: a moon jellyfish (Aurelia aurita), about 25 centimeters across.LEFT: UNIVERSITY OF KANSAS, A. DIAMANT; RIGHT: UNIVERSITY OF KANSAS, P. CARTWRIGHT (VIA EUREKALERT)
Mini jellies
Species: Kudoa iwatai
Genome size: 22.5 million base pairs
The evolutionary origin of a large group of microscopic parasites called myxozoans was confirmed by a study published in PNAS last fall (November 16), and the result is somewhat surprising. Myxozoans are in fact tiny, degenerated jellyfish.
Sequencing the genomes of two myxozoan species at the University of Kansas Medical Center, researchers found that these peculiar parasites evolved from free-living marine animals. With a genome more than 20 times smaller than that of the average jellyfish, myxozoan species Kudoa iwatai has one of the smallest animal genomes ever reported.
“Because they’re so weird, it’s difficult to imagine they were...
While myxozoans do have small structures resembling the stinging cells—or “nematocysts”—of other cnidarians (jellyfish, anemones, and corals), their genomes lack several components, including Hox genes, previously thought to be essential for animal development.
“Myxozoa absolutely redefines what we think of as animal,” said Cartwright in the statement. “First, we confirmed they’re cnidarians. Now we need to investigate how they got to be that way.”
 The underside view of a gecko walking on a window in JapanWIKIMEDIA, ALPSDAKE
The underside view of a gecko walking on a window in JapanWIKIMEDIA, ALPSDAKE
Gecko gymnastics
Species: Gekko japonicus
Genome size: 2.55 billion base pairs
The evolution of tail regeneration and super-adhesive toe pads—two of a gecko’s more unusual traits—may soon be better understood thanks to the newly sequenced genome of Gekko japonicus, published in Nature Communications (November 24) by a team from Nantong University in China.
One of the study’s key findings was the expansion of a set of genes associated with beta-keratin, a protein involved in making the tiny hair structures, or setae, lining a gecko’s toe pads. “Our data reveals that the expansion of beta-keratin gene family is essential to the clinging ability of G. japonicus,” wrote the authors in their paper.
The study additionally found evidence in the genome for the adaptive evolution of tail regeneration as part of the gecko’s tail-shedding response to attack by predators. It further linked changes in the opsin gene to the gecko’s sharpened nocturnal vision, and the diversification of olfactory genes to the animal’s sophisticated sense of smell.
The gecko genome is the largest reptilian genome sequenced so far and, according to the researchers, the data are likely to be useful not only for the development of bioinspired adhesives, but also for the study of reptile evolution and diversification.
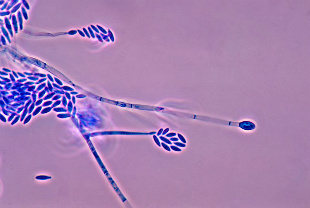 Fusarium verticillioides, a species closely related to F. fujikuroi, was also used in the study.WIKIMEDIA, CDC
Fusarium verticillioides, a species closely related to F. fujikuroi, was also used in the study.WIKIMEDIA, CDC
Subtelomeric subtlety
Species: Fusarium fujikuroi
Genome size: approximately 44 million base pairs
A study comparing newly sequenced genomes from multiple isolates of a fungus infecting rice, published in Genome Biology and Evolution last fall (October 15), has offered up clues about the evolution of diversity in the lineage.
Fusarium fujikuroi, a filamentous fungus, causes bakanae disease in rice and can result in severe crop depletion during outbreaks. The fungus grows inside infected rice plants, stunting normal growth and causing the host to produce empty or partially empty grains.
A team led by investigators at the University of Milan found that many gene sets associated with fungal production of secondary metabolites—such as pigments and toxins that aid infection without being directly involved in development—were concentrated in the subtelomeric regions of DNA at the ends of chromosomes.
Comparing the sequence with available genomes of closely related species also showed that the subtelomeric region contained much of the variation between isolates, suggesting a key role for this region in the fungus’ pathogenic lifestyle. “Focused generation of genetic diversity in subtelomeric regions may thus be particularly important for allowing the fungus to rapidly adapt to changing environments and host colonization,” the authors wrote in their paper.
 An amelanistic corn snakeWIKIMEDIA, TJP2000
An amelanistic corn snakeWIKIMEDIA, TJP2000
White snakes
Species: Pantherophis guttatus
Genome size: approximately 2 billion base pairs
A team from the University of Geneva has sequenced the corn snake genome, according to a paper published in Scientific Reports last fall (November 24). As one of a handful of reptiles to have had their genomes sequenced, the corn snake joins what researchers call a “restricted group” of model species for the study of reptilian evolution.
“This species is perfect for investigating the development and evolution of reptiles because it breeds easily, it is oviparous, and non-venomous,” study coauthor Michel Milinkovitch of Geneva said in a press release.
Sequencing about 85 percent of the snake’s genome, researchers were able to locate the cause of amelanism (or albinism) in the species. Amelanistic snakes, which have pale or white skin where normal snakes are darkly colored, lack a functional copy of the OCA2 gene, the researchers found. The gene codes for a receptor located in the membranes of melanosomes.
Milinkovitch said in the press release that the new data will provide a useful tool to study the genetic determination and evolution of skin color, a feature that is “particularly important for the camouflage of these animals in their natural habitat.”
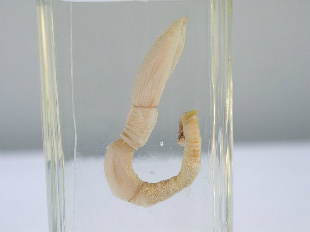 Preserved acorn wormWIKIMEDIA, NECROPHORUS
Preserved acorn wormWIKIMEDIA, NECROPHORUS
Deuterostome duo
Species: Saccoglossus kowalevskii and Ptychodera flava
Genome sizes: approximately 1 billion base pairs each
Deuterostomes are a large taxonomic group of animals consisting mainly of echinoderms (such as sea stars) and chordates (including all vertebrates). But a study published in Nature last fall (November 18) has provided fresh insight into the ancient beginnings of this group with the newly sequenced genomes of two species of acorn worm, reclusive burrowing creatures that live on the seabed.
“Acorn worms are our most ancient deuterostome relatives, dating back to the origin of deuterostomes, around 570 million years ago,” coauthor Oleg Simakov of the Okinawa Institute of Science and Technology Graduate University wrote in an email to Live Science. “The genomic data fills in the gaps in our understanding of their evolution.”
Simakov and his colleagues found that acorn worms share 8,716 gene families with other deuterostomes. In particular, the study highlights genes relating to the formation of gill slits in the worm that could be linked to the development of the pharynx in humans and gills in many other deuterostomes.
Interested in reading more?





{kind=link}
{kind=link}
{kind=link}
{kind=link}