Immune System vs. Cancer
The comeback of an old idea in immunology prompts a rethink of cancer progression and approaches to treatment.

In a photo I often use in presentation slides of my work, I’m standing in nothing but my swim trunks and sunglasses, covered in mud up to my ears. Next to me is my long-time colleague Joe Trapani, in much the same state, with a raccoon-like pattern on his face where he has smeared the mud away. We’d just spent an hour lounging in the Israeli side of the Dead Sea, the deepest hypersaline lake in the world, full of rich mineral mud that attracted visitors for thousands of years. I explain to my audience that we’re grinning like fools in the photo in part because of the way we looked but also, perhaps, because we were feeling so clever for coming up with...
We had just come from the 1993 EMBO Cell-Mediated Cytotoxicity meeting in Jerusalem where a Swiss group, led by Hans Hengartner, reported the phenotype of a knockout mouse lacking the ability to produce the pore-forming molecule, perforin. Released by the killer cells of the immune system, perforin damages target cells or pathogens by punching holes in their plasma membranes. I had worked on the transcriptional control of perforin during my postdoctoral studies at the National Cancer Institute (NCI) in Frederick, Md., in the late 1980s. Joe, then at Memorial Sloan-Kettering Cancer Center in New York, had studied the perforin gene and attempted to make his own perforin knockout mouse. The two of us listened to the presentation with rapt attention, with the same thought: might this model system reveal a role for perforin in early cancer elimination by immune cells?
With their mouse model, Hengartner’s group demonstrated how fundamentally important perforin was to the immune system’s ability to kill cells infected with bacteria or viruses. While most of the attendees were captivated with the immunological implications, such as which killing pathways remained in the absence of perforin, we wondered whether the model might also uncover cell-mediated killing of tumors, a process called immunosurveillance. Joe and I reasoned that if cancer cells are regulated via perforin-releasing lymphocytes, then mice lacking perforin should develop more cancers than wild-type mice with normal perforin function.
The idea that the immune system could survey the body for cancerous cells, killing the ones that looked abnormal, was not novel. It was suggested in passing by German scientist Paul Ehrlich at the turn of the 20th century, long before even rudimentary understanding of the immune system existed. Fifty years would pass before scientists had experimental evidence to support the idea. Even then, many doubted that the immune system was capable of detecting cancer. After all, tumors originate from our own cells, which the immune system is trained to ignore.
The first formal demonstration of the immune system’s ability to “see” cancer came with the discovery of tumor-specific antigens in the 1950s and 1960s. Lloyd Old, George Klein, and others used tumor transplantation models to show that cancer cells indeed express specific antigens that can be recognized by the immune system as different from healthy cells. In the 1960s, one of my most famous fellow countrymen, Frank Macfarlane Burnet, along with the American Lewis Thomas, drew upon their own research, as well as Old and Klein’s findings, to propose the term “cancer immunosurveillance.” They envisioned it as a process by which the immune system recognized and destroyed cancer cells very early in cellular transformation. Although this hypothesis was formulated without any direct supporting experimental data, it was so compelling that it was enthusiastically embraced by the medical and basic science communities.
In 1973–74, a serious challenge to the cancer immunosurveillance hypothesis arose when Osias Stutman, at Memorial Sloan-Kettering Cancer Center in New York, tested a central prediction of the theory: immunodeficient mice should develop more spontaneous and carcinogen-induced tumors than their wild-type counterparts. In a series of very nicely performed experiments, Stutman could not validate these predictions and his experiments seemingly sounded the death knell for the cancer immunosurveillance hypothesis.
Joe and I were well aware that this wouldn’t be a popular question to resurrect—most researchers had bid good riddance to the idea that the immune system might keep cancer at bay. But, with a healthy dose of youthful curiosity, we waded in.

One reason for my interest in the perforin knockout was my research on natural killer (NK) cells, which produce perforin as part of their killing arsenal. NK cells were first described by Ron Herberman at the NCI in 1975 as cells with a natural propensity for killing tumor cells in vitro (hence their name). No other immune cell had this spontaneous ability. Its effects were impressive: a few NK cells would kill many types of cancer cells in culture without any additional activation. Now I knew perforin was essential. It seemed simplistic to think that cytotoxic lymphocytes, such as NK cells, might behave the same way in the tissues of a live animal, but that was the idea that I wanted to test.
By then a few researchers had realized several flaws in Stutman’s experiments. First, the immunodeficient mice that he had used weren’t actually devoid of NK cells, as he had thought at the time, leaving the possibility that immunosurveillance was intact in these mice. Second, the strain of mouse he used was overly susceptible to the carcinogen and therefore the immune system may well have been overwhelmed by cancer development, failing to reveal immune control.
Coming back to Australia after our trip to Israel, Joe and I started planning a convincing way to test the concept of cancer immunosurveillance using perforin-deficient mice at the Austin Research Institute in Melbourne. Several groups had made perforin knockout mice and Bill Clark from the University of California, Berkeley generously provided his strain. We wanted to cross the perforin knockout mouse with one we could be sure would develop cancer, so we acquired a mouse with a genetic predisposition for cancer—the p53 knockout mouse from Alan Harris at the Walter and Eliza Hall Institute. p53 mutations are found in over half of all human cancers, and mice that lack p53 develop a spectrum of sarcomas, lymphomas and, more rarely, adenocarcinomas over their life span. We decided to intercross mice that had one allele of p53 and one allele of perforin knocked out to generate progeny with nine different genotypes (see graphic). It took more than 50 breeding pairs to generate hundreds of mice. We coded the newborns and simply left them on the shelf of our pathogen-free animal house to age, without breaking the code.

To test for the role of the immune system in cancer detection and elimination, we inbred female and male mice with both a predisposition to cancer (a single allele p53 knockout) and immunodeficiency (a single allele for perforin—an immune cell-secreted molecule that kills pathogen-infected cells). The progeny of these breeders had nine different genotypes. All mouse genotypes lacking both perforin alleles developed cancer sooner than those with one or two intact perforin genes, showing for the first time that immune-mediated killing was involved in cancer surveillance and elimination.
Our greatest concern was whether we could age sufficient numbers of mice long enough to obtain statistically significant data, given that no one had reported aging the perforin-deficient mice beyond a year. Thus we obsessively watched these mice for signs of cancer development, keeping fingers crossed that no facility disaster would compromise our significant investment. Remarkably, some of the oldest mice in this study developed such a relationship with Kevin Thia—one of the PhD students monitoring them—that no one else could go near them without fear of being bitten!
Along the way we sacrificed mice that exhibited lumps, weight loss, or other signs of disease. Within the first 200 days, a number of mice fell ill, but after determining their genotype we saw that, as expected, all of these mice lacked both copies of p53. Interestingly, mice that lacked both copies of perforin and p53 developed lymphomas significantly sooner than the p53 double-knockouts that had functional perforin. This excited us, but we anticipated that the mice missing only one p53 allele would be even more revealing since the cancer development in these mice was slower, allowing any involvement of immunity to become more obvious. It was not until 600 days into the study that we had decoded and genotyped enough mice with associated disease to appreciate two remarkable facts. One, mice with one working copy of p53 and lacking perforin developed spontaneous B-cell lymphoma earlier and more frequently than their littermates with at least one perforin allele intact. Two, it did not matter whether the perforin knockout mice had one or both working copies of p53—both developed B-cell lymphomas at the same rate, making us wonder why we had bothered using the p53 knockout in the first place.
Joe, Kevin, and I were excited and relieved that perforin was an unequivocally important suppressor of B-cell lymphoma. We had gone some way to validating Burnet’s hypothesis and the study was the catalyst for many others of similar ilk. While these studies were ongoing, we started a collaboration with Dale Godfrey at the University of Melbourne to assess the importance of NK cells and a small population of specialized T cells, called NKT cells, in immune surveillance of cancer. Rather than using p53 mutant mice, we were studying a mouse model of fibrosarcoma induced by methylcholanthrene (MCA), a carcinogen component of the tar found in cigarettes. We showed that mice lacking either NK or NKT cells were more susceptible when exposed to the carcinogen than the wild type.1 This work was the first to clearly demonstrate a role for early (innate) immunity in control of carcinogen-induced tumors.
Seven years after that pivotal meeting in Israel, Joe and I submitted and published our perforin knockout aging study.2 At the onset of this first aging study, he and I relocated to the Peter MacCallum Cancer Hospital in Melbourne and I began to develop an interest in other death-inducing pathways that lymphocytes might use to suppress tumors. Many other experiments were underway in my new lab, but this publication was my first major breakthrough in this area and complemented a lot of in vitro work that Joe and I had published in the intervening years.

Our 2000 publication in the Journal of Experimental Medicine did not go unnoticed. Shortly afterwards, I was invited by the director of the Ludwig Institute, Professor Lloyd Old, to speak at the International Symposium of Cancer Immunosurveillance in New York, which was quite an honor. Lloyd has devoted his career to promoting the discipline of cancer immunology, and at that meeting he introduced me to many key people in the world of tumor immunology.
No one opportunity was more important than meeting with Robert Schreiber, who seemed to be the only other person in the world prepared to undertake long-term aging experiments of the kind necessary to characterize cancer immunosurveillance. Bob had a distinguished history in cytokine signaling, particularly with the inflammatory mediator, interferon gamma (IFNγ). In broadening my interest in cancer immune surveillance, I had noted Bob’s work on interferons, including a landmark paper in 1998 that largely supported the idea that IFNγ could protect the host from cancer development, much the same way that perforin did.3 On meeting, we appreciated the great opportunity that existed for us to join forces. For a few years, we each independently completed studies that further supported the cancer immune surveillance hypothesis.
Clearly, despite the immune system’s survey and rejection of cancer cells, tumors still develop in the presence of a functioning immune system. To account for this, Bob and his colleagues refined the concept of cancer immune surveillance into what they called “cancer immunoediting.” The origins of this term immunoediting came from his laboratories’ 2001 study published in Nature.4 They showed that tumors induced by carcinogens in wild-type mice were less immunogenic—less likely to spur an immune reaction—than those derived from immunodeficient RAG knockout mice that lack all T and B cells. They proposed that the tumors from the RAG-deficient mice were more immunogenic because presumably they had not been sculpted by T and B cells as the tumor developed, whereas the wild-type tumors had.
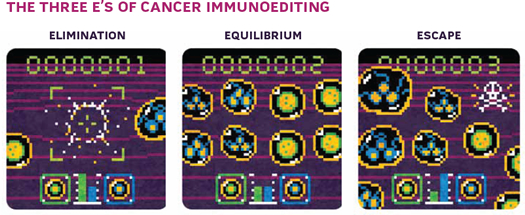
To explain this, Bob proposed three phases of immune system–tumor interaction: elimination, equilibrium, and escape. The elimination phase is essentially immune surveillance, in which the immune system detects and destroys tumor cells. Tumor cells that aren’t destroyed can enter a state of equilibrium with the immune system, in which the growth of the tumor is checked by immune cells killing a proportion of the cancer cells. During this period, however, tumor cells may continue to accumulate mutations, potentially generating variants that resist, avoid, or suppress the antitumor immune response. This leads to the escape phase, in which the tumor growth is unchecked by the immune system. Cancers that doctors see in the clinic have achieved this third phase.5

Bob based his theory on a number of observations. Still, while there was evidence for surveillance/elimination and escape phases, there was no real characterization of the equilibrium phase. Clinically, tumors can persist for years without developing into full-blown cancer; there have been cases of cancer inexplicably going into remission for many years. The equilibrium process was inferred largely from these clinical observations. Most striking were the reports of organ transplant recipients developing cancer that originated from the donor, despite the donor’s having been healthy and apparently cancer free. In such cases, it is possible that the tumor was being held in the equilibrium phase in the donor, in a dormant state, and that transplantation of the organ into an immunosuppressed host allowed tumor outgrowth. Although several mechanisms underlying tumor dormancy have been proposed, there was very little evidence that the immune system played an important role. We wanted to develop a mouse model that mirrored the experience of patients whose cancer resurfaced after many years of lying dormant.
By around 2002, Bob’s group and ours were doing similar experiments in a mouse model of fibrosarcoma induced by methylcholanthrene (MCA). We had both noted that some mice treated with a limiting dose of this powerful carcinogen did not develop clinically apparent cancer but rather harbored tiny stable lesions that eventually disappeared. We realized that these mice might provide a model to test the concept of equilibrium. Bob’s lab began injecting large cohorts of 129 mice with low doses of MCA, and shortly thereafter we commenced an identical experiment in a different background strain of mouse (C57BL/6). Mice that developed progressively growing tumors were removed from the study, leaving mice that had small, nongrowing masses at the site of the injection. We saw that these “equilibrium phase” tumors were infiltrated with immune cells. When we depleted mice of either strain of T cells and removed the protective IFNγ with antibodies, the stable lesions rapidly progressed into aggressive cancers. Surprisingly to me, when we removed NK cells with antibodies, the small cancers did not progress. It suggested that NK cells, which had been important in the elimination phase, were not involved in keeping tumors in equilibrium.6
There is clearly much more work to be done in this mouse model of cancer, and in other models. Why do some tumors enter equilibrium and others do not? What genetic signature does a tumor have to have in order to enter equilibrium? By what mechanism does the immune system suppress a tumor’s outgrowth? It is also unclear whether this state actually exists in human patients, and if so, how this knowledge could be used to develop new approaches to improving patient outcomes. Instead of trying to remove all vestiges of cancer, might it be possible to confine the cancer to a state in which the immune system controls it?
Other than elevated cancer incidence in transplant recipients receiving immunosuppressive drugs, evidence of cancer immunoediting in humans is comparatively sparse. However, recent studies of human cancers suggest that the type of immune cells present in and around cancer predicts the outcome of the patient. Positive outcomes are correlated with the presence of activated effector cells, whereas the presence of the immune suppressing T regulatory cells are associated with a poorer prognosis. This strengthens evidence for the role of the immune system in cancer, but more evidence is needed.
Our work has taken many years. That is the price paid to study cancer in a biologically relevant context, where you look at tumors arising over the lifetime of a mouse. This has distinguished our work from others that have relied on short-term transplantable tumor models. However, some scientists rightfully argue that inbred mice don’t capture the complexity of the human disease and their cancer predisposition is low compared with humans. Lacking an alternative, we still rely on our mouse models to reveal new ideas about cellular and molecular mechanisms of disease.
Joe Trapani and I felt quite clever to attack a question that helped rekindle the debate over immune surveillance. Twenty years later, there are spots of clarity, where we’ve cleared the mud away, but the picture is still a bit blurred. n
Interested in reading more?




