

It was the spring of 2009; US President Barack Obama was just settling into the White House when the Centers for Disease Control and Prevention (CDC) informed him that a new influenza A virus had suddenly appeared in kids in Southern California and was likely to spread around the world. The global pandemic that scientists feared had finally arrived, only no one predicted it would come from North America.
National security advisors and public health officials had been warning the White House for years that dangerous flu viruses were circulating in birds that were only a handful of mutations away from jumping to humans, potentially sparking a pandemic like the notorious 1918 influenza outbreak that claimed more than 20 million lives globally. But US health officials expected the next globe-trotting virus to arise in Asia, the hotbed of zoonotic pathogens such as H5N1, or “bird flu,” and the severe acute respiratory syndrome (SARS) virus, which nearly caused a pandemic in 2003. So they were caught off guard when cases of a new disease started appearing in children in San Diego in April 2009. Officials were doubly surprised when the virus was identified as a swine influenza. All three influenza pandemics of the 20th century had been bird strains.
Even more baffling, the virus had an unusual genome, with pieces derived from three different swine influenza lineages, including a Eurasian lineage not previously observed in the Americas. As the new virus spread around the globe, sickening millions and killing some 200,000 young, otherwise healthy people, Obama’s advisors had no answers for how the unusual virus had evolved and where or when it jumped from animals to people. After diagnostic testing rolled out globally, it appeared that the epicenter of the pandemic had been in central Mexico.
Rumors began to circulate that the virus, which was spreading rapidly among humans through contact and aerosols, was engineered by human hands. The CDC scoffed, but the World Health Organization (WHO) announced that it was seriously investigating a memo sent to the organization by Australian biologist Adrian Gibbs, who laid out his evidence for why the virus was not of a natural origin but rather had leaked from a laboratory performing genetic experiments for vaccine production. Gibbs was considered a credible scientist who had participated in the development of the Roche anti-influenza drug Tamiflu. Scientists and public health officials shuddered. A lab-error pandemic virus was the last thing the world needed. Years of progress against vaccine-preventable diseases had already been undone by British doctor Andrew Wakefield’s falsified autism-MMR link, which would not be retracted for another year, fully 12 years after its publication.
Scientists cannot create data out of thin air every time there is an outbreak.
Soon, though, two seminal studies soundly refuted the lab-leak theory and revealed how the virus arose naturally in pigs. The detailed evolutionary history spanning several decades was more than we had learned about the animal sources of most other zoonotic outbreaks. Many considered the book on the pandemic’s origins was effectively closed.
Still, some bits of the story didn’t add up—notably the prevailing notion that the virus came from pigs in Asia. I had recently joined the National Institutes of Health (NIH) to study influenza in humans, but the pig story piqued my interest. So I switched to researching pigs. Dropping human research to study swine might seem like an ill-advised career move, but I had an unusual amount of freedom at the NIH. I wasn’t living grant-to-grant like most epidemiologists, so I could follow obscure hunches.
It ultimately took our international teams seven years to track the pandemic’s origins to an unstudied corner of pig farming. Along the way, I learned that raising a pig—and a pandemic—is more complicated than I imagined. Still, tracing the origin of a pandemic, even long after the fact, can be done using genetic data, and the full understanding of how a disease emerged is worth the years of sleuthing.
Quashing the “lab leak” rumors
While in retrospect it’s easy to be glib about the idea that the 2009 virus leaked from a laboratory somewhere, Gibbs pointed out some features of the virus that were important to explain. He doubted the virus came from nature because no genetically similar influenza viruses were known to circulate in pigs anywhere in the world. As a result, the virus sat on a “long branch” of the influenza phylogenetic tree, separated by many mutational differences from other swine flu viruses. Also, the virus’s proteins had an abundance of lysine residues. He and his colleagues noted that standard vaccine production practices of growing influenza viruses in chicken eggs—a method still used by the majority of flu vaccine developers—can lead to an increase in lysine residues, so Gibbs concluded that the newly emerged swine flu had been propagated in a laboratory, likely as a part of vaccine-related research.
This idea did not hold up long against scientific scrutiny. All five of the WHO’s collaborating influenza centers evaluated Gibbs’ claims, and all arrived at the same conclusion: there was no reason to believe the virus was unnatural. Human, avian, and swine flu viruses naturally differ somewhat in lysine composition, and the amount of lysine seen in the strain responsible for the 2009 outbreak was consistent with natural increases seen in swine flu viruses historically. The genetic distance, too, proved unremarkable.

Scientists had a wealth of genetic data from swine influenza viruses to compare against, as veterinarians have routinely performed diagnostic testing, sometimes sequencing entire viral genomes, to combat flu and monitor viral trends. The NIH’s public repository GenBank contained more than 500 genetic sequences from swine influenza viruses collected in the US, Canada, and multiple countries in Europe and Asia at the time of the 2009 outbreak. And that data revealed many swine flu viruses are somewhat isolated on the phylogenetic tree—the result of insufficient sampling from certain time periods or locations.
Research teams from the US CDC and the University of Hong Kong used these genetic data to catalog the natural evolutionary origins of the virus in fine detail in studies published in Science and Nature, respectively. Both studies showed that the pandemic virus’s genome belonged to the H1N1 subtype, which is found in swine globally, and concluded that the virus arose through natural processes. But an unresolved question was where.
The exact origins of the 2009 virus were difficult to pin down because flu virus genomes are composed of eight individual segments that can be swapped in their entirety during replication when two “parent” viruses co-infect a single host cell. This creates genetically new offspring in a process known as reassortment. And the 2009 virus epitomized why swine are called “mixing vessels” for avian and human strains: the eight segments of the pandemic virus’s genome had notably different evolutionary histories connected to three geographically distinct swine flu lineages, some of which were introduced into pigs from birds and others from humans.
Two genome segments came from a Eurasian swine H1N1 lineage that jumped from birds to pigs in the 1970s and remains prevalent in pigs in Europe and Asia today. One genome segment was derived from a classical swine H1N1 lineage that first emerged in US pigs during the 1918 flu pandemic and continues to circulate in the US and Canada, with occasional incursions into Asia. The remaining five segments were derived from the “triple-reassortant” lineage that emerged in US pigs in the 1990s and was itself a mix of avian, human, and classical swine influenza A viruses. Triple reassortant viruses are established in the US and Canada, but also have been introduced into Asia.
In their Nature paper, the Hong Kong group provided data compatible with an Asian origin. They had found new swine flu viruses in abattoirs in Hong Kong that resembled the 2009 H1N1 pandemic more closely than any other swine virus known globally—viruses whose genomes were mixes of segments from the same three swine flu lineages as the pandemic virus. Although none contained the exact same segments as the newly emerged pathogen, the authors noted that all three major genetic components of the virus were found in swine in Hong Kong, which routinely receives pigs from mainland China. So, it seemed reasonable that with additional sequencing in Asian swine, a perfect segment-by-segment match would eventually show up. After all, China hosts half the world’s swine population and only a tiny slice had been sampled for flu. Plus, the genetic sequences of the flu viruses from Hong Kong’s pigs were more closely related to the pandemic virus in terms of mutational distance than viruses sampled from pigs elsewhere. The paper included strong caveats, especially with regard to sampling, but an Asian-origins scenario was easy to accept because it fit with Asia’s historical role as the source of H5N1 “bird flu,” SARS, and two previous influenza pandemics (in 1957 and in 1968), and matched where scientists presumed the next pandemic would arise.
Still, there were lingering gaps in the story that bugged experts in the field, myself and the Nature study authors included. Where the new virus overlapped with Asian sequences, there were mutational differences that suggested many years of unobserved evolution. What was the virus up to during that time? Plus, from a clinical perspective, the first major outbreak in humans occurred in North America. Weeks before the flu was first identified in California, clusters of an unusual pneumonia appeared in Mexico. The viral sequences obtained from patients in Mexico were genetically diverse, suggesting the virus had already been circulating there for many weeks, possibly months. No scientist could explain how a virus that appeared to arise in Asian swine could trigger an outbreak in humans in Mexico without setting off similar transmission chains in Asia. And no one had found an animal with the exact same virus strain. That is to say, there was no “smoking pig.”
A History of Viral ReassortmentThe genome of the H1N1 virus that jumped to humans in 2009 included two segments from a Eurasian lineage, one segment from a classical lineage, and five segments from the “triple-reassortant” lineage, itself the product of viral intermixing. 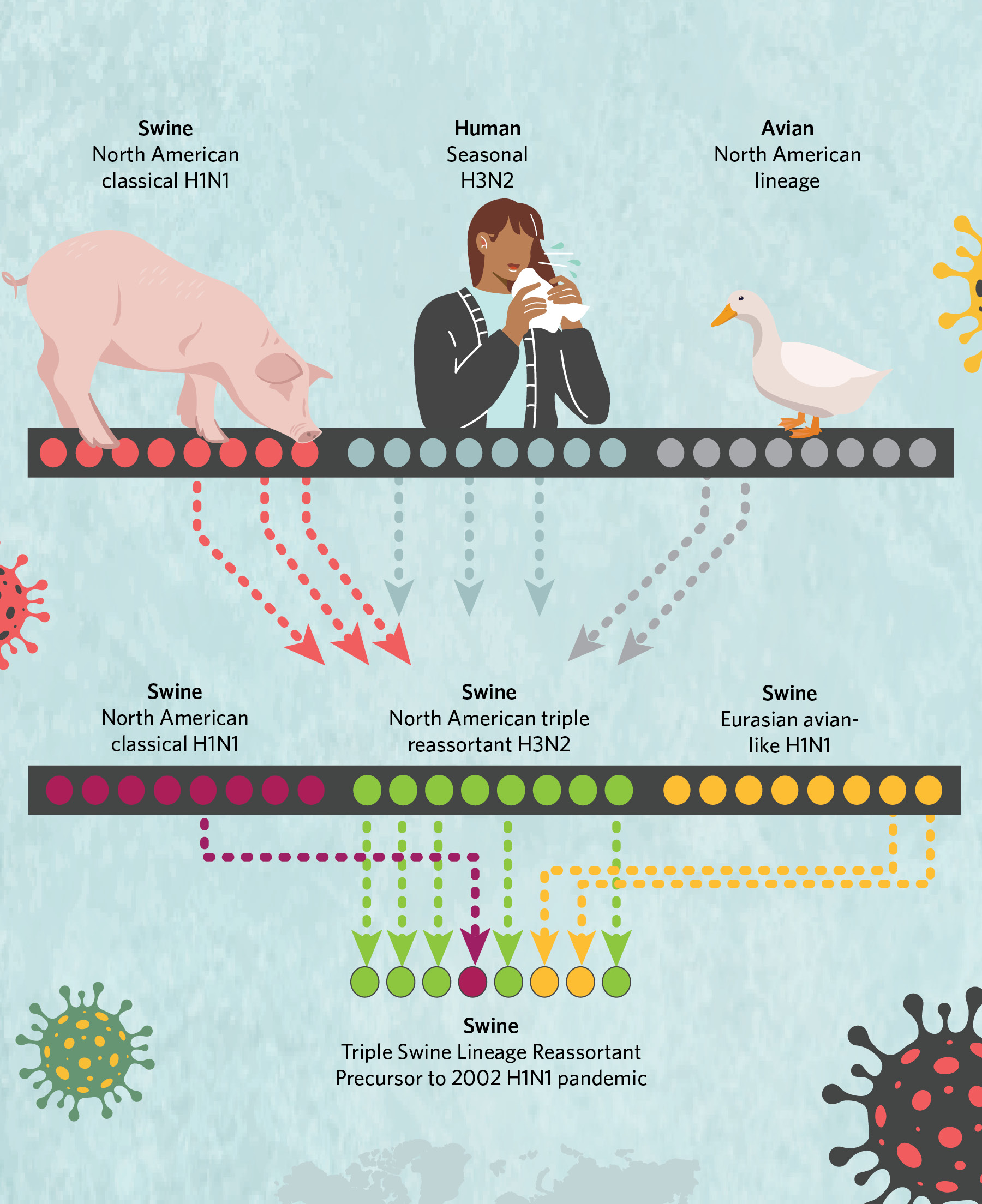 © istock.com, bgblue; © istock.com, ankomando; © istock.com, Julie_fdt; © istock.com, Irina_Strelnikova |
The hunt for the smoking pig
Interest in the pandemic waned after cases fell during 2010. But the question of the virus’s origins remained a topic of discussion among a small group of pathogen hunters and evolutionary biologists who met informally in Leuven, Belgium, a small Flemish city renowned for its beer, chocolate, and one of Europe’s leading universities. In a building marked for demolition, we sketched out on a whiteboard possible scenarios for the pandemic’s origins, each of which seemed far-fetched. Could “patient zero” be a Chinese hog farmer who flew to Mexico? Exactly how many people from China’s swine-farming regions fly to Mexico each day? Or did an infected Asian pig make its way to Mexico some time before the pandemic? Do pigs ever fly from China to Mexico? No one knew.
One silver lining of the 2009 H1N1 pandemic was an increase in funding for research and surveillance of influenza viruses in pigs on a global scale, allowing myself and other scientists to dig deeper into the many unknowns about influenza in pigs. During the pandemic, the US Department of Agriculture (USDA) established routine surveillance and genomic sequencing of flu viruses collected in US herds. The UK Animal and Plant Health Agency established a research consortium to characterize virus diversity in European swine herds. Numerous research groups published their countries’ first surveillance reports for influenza in swine, even in Brazil and Australia where the herds were considered influenza-free. It turned out that anywhere there were pigs, there was flu. Swine veterinarians in many countries suddenly became interested in teaming up with me and my collaborators to figure out the genetic makeup of these newly discovered viruses and to develop customized vaccines.
Impugning a single country or animal species oversimplifies a geographically and ecologically intermixing world.
Some of the flu viruses identified by these efforts were genetically similar to the Eurasian viruses found in Europe or the triple-
reassortant and classical virus lineages circulating in the US, indicating that the viruses had traveled from centers of swine production in Europe and the US to other countries that regularly imported their live swine. Transporting live pigs across oceans is expensive and cumbersome, but many countries were rapidly modernizing their swine production towards the end of the 20th century and needed sows bred to produce larger litters of piglets, which were only available from the US, Canada, and Europe. Imported swine had to be documented to be free of certain pathogens such as African swine fever, but most pigs were not tested for influenza or quarantined. Because influenza frequently infects pigs asymptomatically, it makes an excellent stowaway.
How had influenza viruses reached geographically isolated areas such as Western Australia, with its stringent quarantine requirements for imported animals? We were learning that the story of the 2009 pandemic had a twist. Yes, one H1N1 influenza virus had managed to jump from a pig to a human to spark the 2009 pandemic. But humans also transmit influenza viruses to swine in what’s known as reverse zoonosis. All that surveillance of flu viruses revealed that, following the pandemic, humans transmitted the virus back to swine all over the world en masse, including in Australia. Humans may refer to the 2009 pandemic as “swine flu,” but from the pigs’ perspective, humans are the disease vectors.
At first, the introduction of human viruses into swine herds created a disease problem largely restricted to pigs. But in each country, the human-sourced pandemic viruses quickly reassorted with other endemic swine viruses to create novel strains, some of which began to infect people.
For example, during the summer of 2012, more than 300 American children, primarily in Ohio and Indiana, were infected by viruses from their show pigs, and my colleagues and I found that the viruses involved in that outbreak contained a piece of genetic material introduced into US herds by humans during the pandemic. This later round of zoonotic infections never established human-to-human transmission, so the strains never went global. But the scare was a wake-up call that new infectious diseases can arise at any time from unexpected sources—even from America’s wholesome state fairs—and that the risk of another influenza pandemic arising from pigs had only increased since 2009.
Still, the origins of the 2009 pandemic remained mysterious—and, frankly, a bit confusing. My collaborators sequencing flu viruses from Latin American pig herds turned up plenty of new viruses of human origin, but none contained genetic material from the Eurasian lineage that had donated two crucial segments to the pandemic virus. The more countries in the Americas that failed to unearth a match to the pandemic virus, or even a shred of evidence that Eurasian viruses had made it there, the more doubtful I became that the pandemic could have originated in the West. Had it come from Asia after all?
The Investigation into H1N1’s OriginsIt was a long journey to figure out what led to the 2009 H1N1 pandemic, but eventually the work not only solved the pandemic’s origins, but also yielded results that should serve as a warning sign: we do not have the surveillance infrastructure in place to track fast-moving and intermixing pathogens inadvertently shipped around the world as livestock operations grow to support an expanding human population. Here are some of the major stops on our journey to tell the pandemic’s origin story. 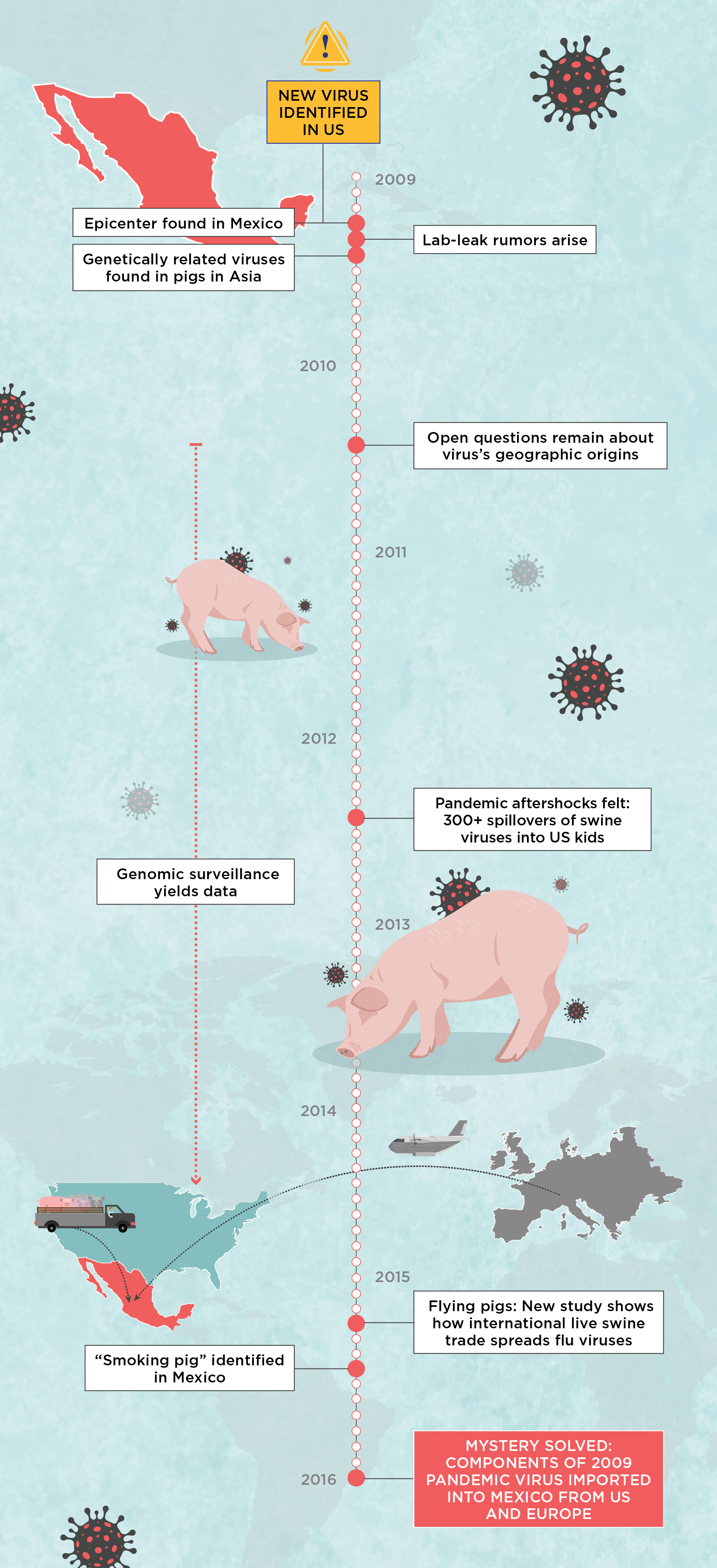 |
Pigs can fly
Even though I found no support for my hypothesis about the pandemic’s swine origins being in the West, six years of digging had not been wasted. By 2015, labor-intensive viral collection, diagnostic testing, lab prep, genetic sequencing, and genetic analyses had revealed how the world’s billion swine move within and between countries, spreading flu viruses efficiently.
We also discovered that most routes are not round-trip. Every pig that legally crosses international borders is reported to the United Nations Comtrade database, yielding data on the numbers of live swine traded between various countries each year. So I collaborated with NIH epidemiologist Cécile Viboud to build simulations from trade data to predict whether a virus residing in one country’s swine population is likely to spread to neighboring countries, or overseas. This simulation, published in 2015 in Nature Communications, predicted that countries such the US that export many pigs act as “sources” for swine influenza around the world. Other countries, including China and Mexico, act as “sinks”—destinations that receive many pigs but where few pigs travel out. In sink areas, imported viruses can take root and evolve into new forms that go undetected without targeted surveillance. In other words, from the perspective of swine influenza, Mexico is like Vegas: what happens there stays there—at least, until it jumps to another species.
Ultimately, this means swine viruses in the US, Canada, and major exporting countries in Europe readily disperse along international trade routes, creating hubs of viral diversity across Asia and in countries such as Mexico. China was not the only country where the classical, Eurasian, and triple-reassortant lineages likely circulated and combined to create new viruses. And, because some of these countries, including Mexico, export few live animals, they could host new viruses for decades that never spread elsewhere. This could create niches for the hidden evolution of viral lineages—which, in theory, could explain the 2009 virus’s novelty and why it was so hard to pinpoint its origin.
And yet, while sampling Mexico’s swine herds in the years following the pandemic did uncover new Mexico-specific viruses, the lineages we found traced back to humans, not Eurasian swine. We saw the same pattern in other Latin American countries. There was no trace of the Eurasian lineage anywhere in the West, likely because there is not much trade in that direction. All evidence pointed toward an Asian origin, I conceded. And that seemed unlikely to change, as research on the 2009 swine flu was slowing as attention turned to more pressing disease threats, including Ebola in West Africa and Zika in the Americas. Funding for flu surveillance in pigs was drying up.
Imported and ReassortedIn early 2009, a virus with an unusual genome popped up in people in central Mexico. It had pieces derived from three different swine influenza lineages, including a Eurasian lineage not previously observed in the Americas. 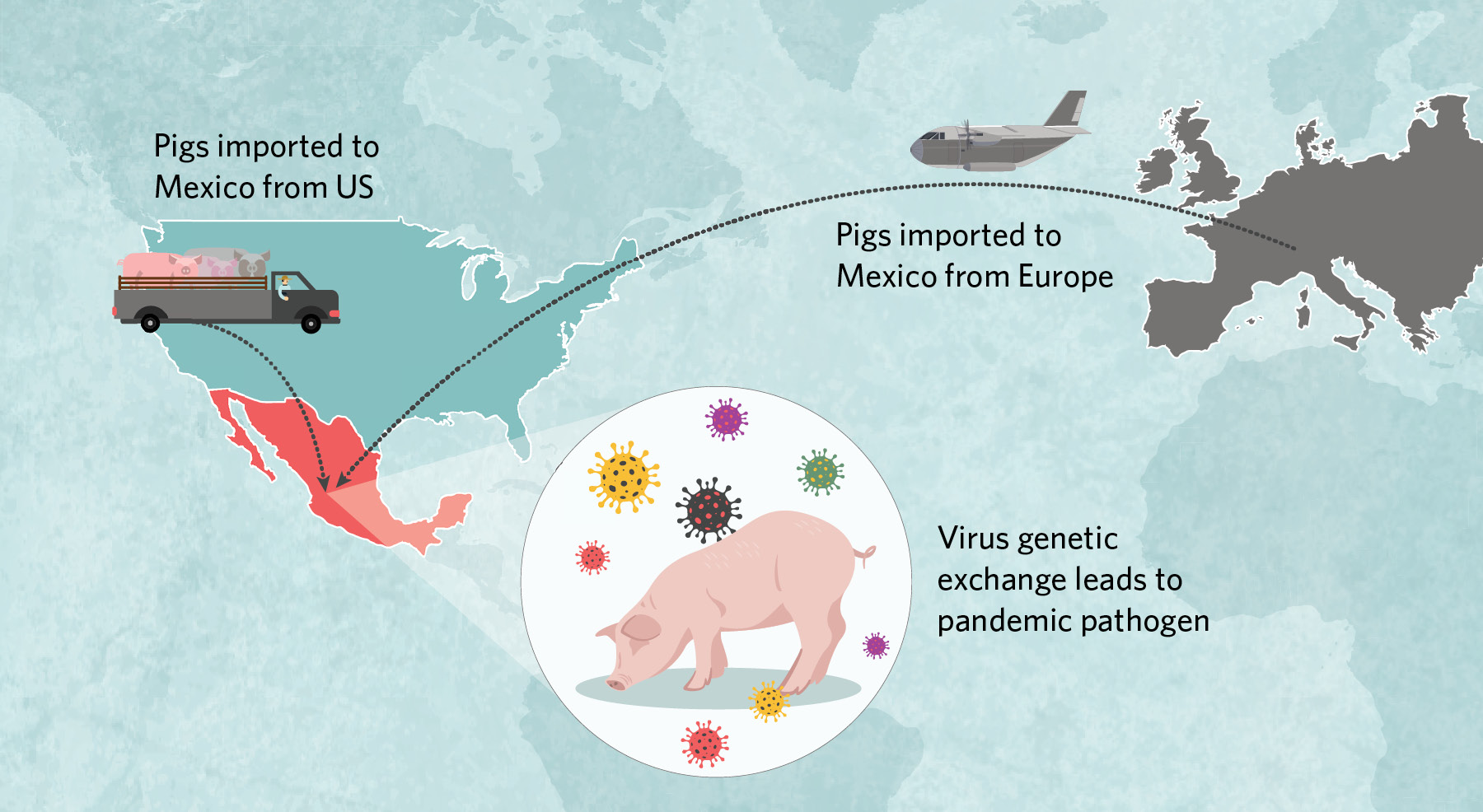 |
Beginning in the 1990s, millions of US pigs were trucked into Mexico, some of which carried classical H1N1 and triple-reassortant H3N2 viruses that then spread across the country’s northern, central, and eastern regions. Around the same time, at least once and possibly twice, Eurasian swine viruses were imported from Europe to central Mexico. There, American- and European-origin viruses exchanged genetic material to create the pathogen that jumped to humans in early 2009. |
The smoking pig
Still, our work on swine influenza continued. The disease remained an issue for US swine farmers, and my collaborator Amy Vincent at the USDA maintained a vibrant surveillance program. So we organized a workshop at her lab in Ames, Iowa, to teach scientists how to analyze genetic data from swine influenza viruses circulating in different countries.
One workshop participant was Ignacio “Nacho” Mena, a virologist who was part of the NIH-funded Center of Excellence for Influenza Research and Surveillance (CEIRS) that Vincent and I also participated in. For the workshop, he brought the genomes of 57 influenza viruses collected from swine by his local veterinarian partners all over Mexico between 2013 and 2015. These genomes were hot off the sequencer, so they hadn’t yet been fully examined. Astoundingly, one of Mena’s Mexican viruses had a gene segment that was related to Eurasian viruses from swine in Europe, seemingly the first documentation of the Eurasian swine H1N1 lineage in the Western hemisphere. The Eurasian segment didn’t match those in the 2009 pandemic virus, but the detection of any genetic pieces of the Eurasian lineage in Mexico meant it was possible that the 2009 pandemic had emerged in North America.
When we looked more closely, Mena’s data turned out to contain 18 smoking pig viruses that matched the 2009 viral lineage segment for segment. Moreover, the 18 viruses were collected from swine in central Mexico, the same region where the earliest clinical signs of a pandemic outbreak had been detected in humans in early 2009. And lastly, phylogenetic analyses placed the Mexico viruses in a clade on the tree that more closely adjoined the clade of human pandemic viruses than any other swine viruses. The pattern was exactly what we would expect if the 2009 H1N1 pandemic originated in pigs in Mexico.
Only one mystery remained: How did Eurasian viruses with a geographical range limited to Europe and Asia wind up in Mexico? US swine veterinarians remained convinced that Eurasian viruses had never reached America’s well-sampled herds. Fortunately, thanks to time-stamped sequences, background data from swine in Europe, Asia, and North America, and a decade of advances in phylogenetic tree-building methods, we were able to reconstruct a detailed history of influenza virus evolution in Mexico dating back to the 1990s. Our findings, published in eLife in 2016, confirmed Mexico’s role as a pig trade sink, and therefore a melting pot for influenza A viruses imported from Europe and the US.
According to the analysis, millions of US pigs were trucked into Mexico along with their classical and triple-reassortant viruses, which over time spread across the country’s northern, central, and eastern regions. Around the same time, Eurasian swine viruses were independently brought into Mexico directly from Europe on at least one, and possibly two, occasions, presumably by an infected sow in a plane’s cargo. The Eurasian viruses had circulated for more than a decade in Mexico undetected, but only in the central region surrounding Mexico City, where the country’s major swine-producing states of Jalisco and Guanajuato are located. Here, American- and European-origin viruses exchanged genetic material through reassortment to create an entirely new pathogen with a hitherto unseen combination of segments that helped the virus jump to humans.
Playing the long game
It took me and my colleagues seven years to piece together how the 2009 H1N1 pandemic happened. What drove us to persist, and continue tracing that virus’s origins even as the rest of the world moved on?
For one, we needed to set the record straight. Although Adrian Gibbs had been wrong about the pandemic coming from a lab, he had laid out questions the scientific community still needed to answer, including why the virus was positioned on a long branch of the phylogenetic tree and where the virus had been hiding out all those years.
It was also worth emphasizing that uncovering the complete evolutionary history of the 2009 pandemic did not pin blame on a single country. Although Mexico was the proximal source of the 2009 pandemic, all the viral components were imported from pigs in the US and Europe. The deeper we dove into the complexity of pathogen emergence in a globalized world, the harder it became to point fingers.
There is also the lingering idea that birds are the only important source of flu; they’re not. Novel flu lineages have been detected in all sorts of animals, most recently in dogs, bats, and cattle. Even we humans play a role; people transmit hundreds of influenza viruses to pigs for each virus that successfully jumps from pigs to humans. If there’s another pandemic swine flu in the future, it will be because humans repeatedly restock pigs’ viral gene pool with new genetic diversity that combines with other pig viruses.
Really, it shouldn’t have come as such a surprise that the first global pandemic of the 21st century came from pigs. For most of the 20th century, influenza was not a problem for pig farmers. But as agricultural production of pigs expanded and modernized at a blistering pace, with backyard farms being displaced by large commercial operations all over the world, the opportunity for viral intermixing grew exponentially and cases of flu in swine began to climb.
Drug companies have tried to manufacture vaccines for swine flu, but they struggle to keep pace with a moving target. Influenza simply evolves too fast and has too many different strains. As the NIH and other funders invest in improving influenza vaccines for humans that broadly protect against diverse strains, it is worth testing out new candidates in pigs, where the problem of matching vaccine and field strains is even more dire. The risk of another pandemic coming from pigs only grows each year.
Of course, the next globe-trotting influenza virus won’t necessarily come from swine. It’s impossible to predict the origin of the next pandemic flu, as scientists struggle to keep up with the effects of globalization, climate change, and economic development in an unpredictable and ever-changing landscape for disease emergence. Impugning a single country or animal species oversimplifies a geographically and ecologically intermixing world. As we enter the third year of the COVID-19 pandemic, to prevent future pandemics, there is an urgent need for scientists from different countries to work together and share data on emerging pathogens.
There is value in being able to explain the animal origins of a new disease right at a pandemic’s onset, not seven years later, to reduce fear and avert misdirected mitigation efforts, for example culling of pigs in 2009. But scientists cannot create data out of thin air every time there is an outbreak. Tracking the global diversity and movement of animal diseases in real time requires ongoing investment and infrastructure. Some countries like Australia already do rigorous testing and quarantine, but most do not. Currently, there is no requirement for animals being shipped around the world to be tested for flu.
We hope our small success emboldens efforts to uncover the zoonotic origins of other novel viruses, even if it takes years of digging. Tracing the earliest chains of human infections becomes near impossible for a respiratory virus like flu or SARS-CoV-2 that causes so many asymptomatic infections and transmits so rapidly. Fortunately, remarkable advances in genomic sequencing and phylogenetic reconstruction can extract hidden signals from animal data going back decades, and the origins of a virus can be uncovered many years after a pandemic event by continuing to follow the genetic traces of its descendants still circulating in animals. Eventually, such work forms a full picture of a disease system spanning humans, wildlife, and domestic animals over time and space, unearthing the root causes of pandemics, including animal trade. The potential of DNA to sleuth unsolved mysteries is only beginning to be tapped.
Editor’s note: A previous version of this story without infographics was published online in September 2020. Be sure to check out the animated and interactive versions of the infographics in this story, which appeared in the first issue of TS Digest.