
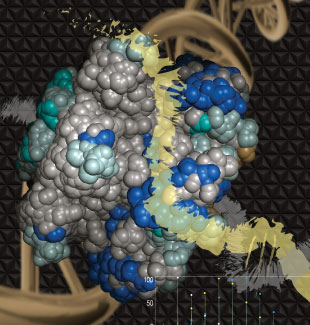
 BINDING PATH: For the archaeon Sulfolobus solfataricus’s mini chromosome maintenance helicase (SsoMCM), high-resolution mass spectrometry monitors the rate of deuterium uptake for free protein compared with DNA-bound protein. The differences (shades of blue) can then be mapped back onto the protein structure to identify putative binding paths for single-stranded DNA (yellow) on the exterior surface of the SsoMCM helicase.MICHAEL TRAKSELISMass spectrometry is today’s go-to technology for proteomics research. The technology makes it relatively straightforward to identify and quantify proteins across a range of samples, as well as to detect the posttranslational modifications that so often govern their behavior.
BINDING PATH: For the archaeon Sulfolobus solfataricus’s mini chromosome maintenance helicase (SsoMCM), high-resolution mass spectrometry monitors the rate of deuterium uptake for free protein compared with DNA-bound protein. The differences (shades of blue) can then be mapped back onto the protein structure to identify putative binding paths for single-stranded DNA (yellow) on the exterior surface of the SsoMCM helicase.MICHAEL TRAKSELISMass spectrometry is today’s go-to technology for proteomics research. The technology makes it relatively straightforward to identify and quantify proteins across a range of samples, as well as to detect the posttranslational modifications that so often govern their behavior.
Such studies tend to treat proteins as isolated entities, effectively inventorying them on an organelle, cell, or tissue scale and drawing inferences from the way those tallies change across conditions. But proteins rarely act alone.
Proteins often aggregate into large multiprotein complexes, for one thing. (See “Cracking the Complex,” The Scientist, November 2015.) They also bind to small molecules, lipids, and nucleic acids.
The Scientist asked researchers who have applied mass spectrometry to a number of such interactions for the lowdown on their techniques.
RESEARCHER: Michael Trakselis, Associate Professor, Department of Chemistry & Biochemistry, Baylor University PROJECT: Mapping protein-DNA interactions in the mini chromosome maintenance helicase (MCM) of the archaeon Sulfolobus solfataricus (Sso) APPROACH: Helicases are ringlike DNA-unwinding enzymes through which one strand of nucleic ...












