
 GRAPHIC BY JENNIFER DOUDNA/UC BERKELEYA decade, ago, when researchers started to unravel the function of a system called CRISPR (clustered, regularly interspaced, short palindromic repeats), which is found in bacteria and archaea, they had little inkling that it would lead to a tool that has taken the world of gene editing by storm. In the past year and a half, the method has quickly become the go-to technique for mutating and editing DNA across the animal kingdom: it works in pretty much every cell type tested so far, from human and mouse to zebrafish and fruit fly. It is so easy that not one but two research groups capitalized on CRISPR to individually mutate almost every gene in human cells (Science, 343:80-84, 2014; Science, 343:84-87, 2014). Most recently, CRISPR enabled researchers to engineer monkeys carrying specific gene disruptions, a feat that has been possible for decades in mice but was never before accomplished in primates (Cell, doi:10.1016/j.cell.2014.01.027, 2014).
GRAPHIC BY JENNIFER DOUDNA/UC BERKELEYA decade, ago, when researchers started to unravel the function of a system called CRISPR (clustered, regularly interspaced, short palindromic repeats), which is found in bacteria and archaea, they had little inkling that it would lead to a tool that has taken the world of gene editing by storm. In the past year and a half, the method has quickly become the go-to technique for mutating and editing DNA across the animal kingdom: it works in pretty much every cell type tested so far, from human and mouse to zebrafish and fruit fly. It is so easy that not one but two research groups capitalized on CRISPR to individually mutate almost every gene in human cells (Science, 343:80-84, 2014; Science, 343:84-87, 2014). Most recently, CRISPR enabled researchers to engineer monkeys carrying specific gene disruptions, a feat that has been possible for decades in mice but was never before accomplished in primates (Cell, doi:10.1016/j.cell.2014.01.027, 2014).
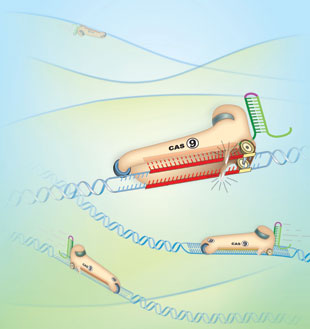
CRISPR functions as a defense system that protects bacterial and archaeal cells from viruses. CRISPR loci in these organisms’ genomes express small RNAs that match sequences in the genomes of invading viruses. When microbes are infected with one of these viruses, CRISPR RNA binds the viral genome through complementary sequence and brings CRISPR-associated enzymes, called Cas, to the viral DNA. The Cas enzymes are nucleases that cut the viral DNA, stopping the virus in its tracks.
The beauty of putting the CRISPR/Cas system to work in other, nonbacterial cells is that it requires just two components: a Cas enzyme to snip target DNA—for example, inside a gene of interest—and an RNA molecule, called guide RNA (gRNA), which binds the target through complementarity. The gRNA is ...














