
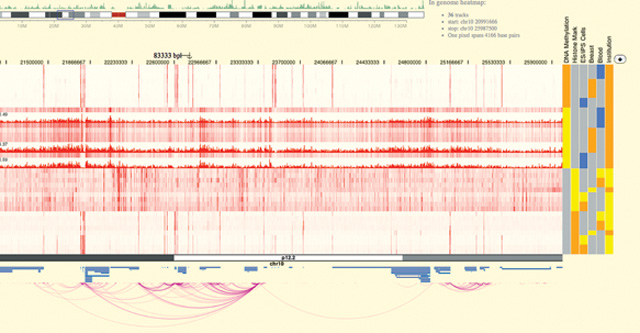
 NEXT-GEN BROWSER: The WashU Epigenome Browser, showing epigenetic data “tracks” in red and long-range chromatin interaction tracks as purple arcs. The tracks are sortable in the heatmap at right.COURTESY OF WASHINGTON UNIVERSITY IN ST. LOUIS
NEXT-GEN BROWSER: The WashU Epigenome Browser, showing epigenetic data “tracks” in red and long-range chromatin interaction tracks as purple arcs. The tracks are sortable in the heatmap at right.COURTESY OF WASHINGTON UNIVERSITY IN ST. LOUIS
September was a monumental month for genome aficionados. The National Human Genome Research Institute (NHGRI)–funded Encyclopedia of DNA Elements (ENCODE) Project released 30 papers in the pages of Nature, Genome Biology, Genome Research, plus another nine in Science, Cell, and the Journal of Biological Chemistry detailing functional features across the human genome. In all, ENCODE researchers performed nearly 1,650 experiments on 147 cell lines assessing transcription, transcription factor binding, chromatin topology, histone modifications, DNA methylation, and more.
The term that encompasses such myriad functional elements is epigenomics, and researchers are now well aware of the importance of such features in development and disease. So much so, in fact, that in 2008, five years after NHGRI launched ENCODE, the NIH funded a second large-scale mapping project. ...














