
 © TETIANA YURCHENKO/SHUTTERSTOCKIn the last decade, a growing number of drug discovery researchers have replaced robots and reagents in their high-throughput screens with computer modeling, relying on software to identify compounds that will bind to a protein target of interest.
© TETIANA YURCHENKO/SHUTTERSTOCKIn the last decade, a growing number of drug discovery researchers have replaced robots and reagents in their high-throughput screens with computer modeling, relying on software to identify compounds that will bind to a protein target of interest.
Researchers often combine virtual screening with other computational tools that make predictions about the activity of individual compounds, such as how they will interact with proteins. Together, these tools help narrow down large libraries of compounds into a subset to test experimentally. The biggest compound libraries boast several million molecules, an unrealistic load for the best-equipped lab to screen the old-fashioned way. Experimentally testing more modest libraries of thousands of molecules would still strain the resources of academic researchers, who are increasingly tackling drug discovery. “As an academic lab, I can’t afford to buy thousands of compounds to do a high-throughput screen, but I could afford to buy 10 or 20,” says Werner Geldenhuys, an associate professor of pharmaceutical sciences at Northeast Ohio Medical University.
Computational tools have their own challenges, however. Depending on the type of predictions the program makes and the size of your library, these screens could take hours to days to run. Some programs require users to perform basic coding. And of course, virtual hits have to be validated in the lab for their ability to actually bind to the target and modulate its activity.
The Scientist surveyed some of the most widely used, freely available computational tools to help you take your drug discovery online.
SHRINKING THE COMPOUND LIBRARY
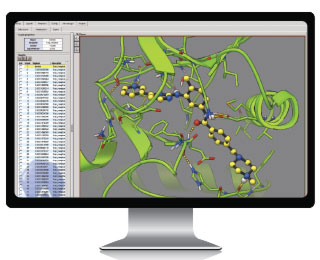 ATTEMPTING TO DOCK: AutoDock programs test and analyze interactions between small drug-like molecules and biological targets. In the AutoDock Raccoon graphical interface shown here, researchers view the 3-D structure of these complexes, such as between the kinase protein (green ribbons) involved in chronic myelogenous leukemia and the anti-cancer drug imatinib (Gleevec). The graphical interface displays the hydrogen-bond network (dotted lines) responsible for the activity of this drug. IMAGE BY DR. STEFANO FORLI, THE SCRIPPS RESEARCH INSTITUTE. vROCSIn traditional high-throughput screening, researchers look through a haystack of compounds to identify a few that bind to a protein target of interest. Screening 20,000 molecules might yield 5 hits.
ATTEMPTING TO DOCK: AutoDock programs test and analyze interactions between small drug-like molecules and biological targets. In the AutoDock Raccoon graphical interface shown here, researchers view the 3-D structure of these complexes, such as between the kinase protein (green ribbons) involved in chronic myelogenous leukemia and the anti-cancer drug imatinib (Gleevec). The graphical interface displays the hydrogen-bond network (dotted lines) responsible for the activity of this drug. IMAGE BY DR. STEFANO FORLI, THE SCRIPPS RESEARCH INSTITUTE. vROCSIn traditional high-throughput screening, researchers look through a haystack of compounds to identify a few that bind to a protein target of interest. Screening 20,000 molecules might yield 5 hits.








