
 © TAMI TOLPA
© TAMI TOLPA
Growing old is a fact of life. And there’s no mistaking it, given the increased fatigue, weakened bones, and ill health that generally accompany aging. Indeed, age is the number one risk factor for myriad diseases, including Alzheimer’s, cancer, cataracts, and macular degeneration. And while researchers are making progress in understanding and treating each of these ailments, huge gaps remain in our understanding of the aging process itself.
“We age so completely and in so many different ways,” says stem cell biologist Derrick Rossi of Harvard University. “We are programmed to die.”
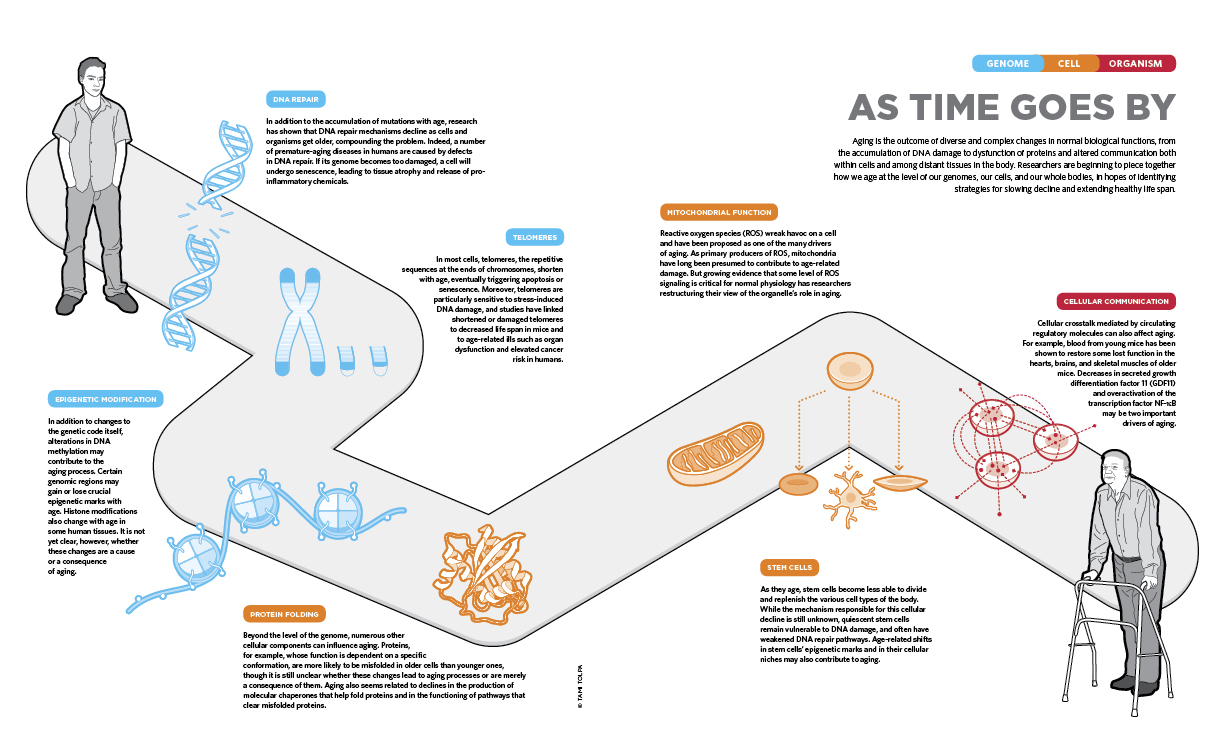
 AS TIME GOES BY: Aging is the outcome of diverse and complex changes in normal biological functions, from the accumulation of DNA damage to dysfunction of proteins and altered communication both within cells and among distant tissues in the body. Researchers are beginning to piece together how we age at the level of our genomes, our cells, and our whole bodies, in hopes of identifying strategies for slowing decline and extending healthy life span.
AS TIME GOES BY: Aging is the outcome of diverse and complex changes in normal biological functions, from the accumulation of DNA damage to dysfunction of proteins and altered communication both within cells and among distant tissues in the body. Researchers are beginning to piece together how we age at the level of our genomes, our cells, and our whole bodies, in hopes of identifying strategies for slowing decline and extending healthy life span.
See full infographic: JPG | PDF© TAMI TOLPAThe aging process can be traced down to the level of cells, which themselves die or enter senescence as they age, and even to the genomic level. Accumulation of mutations and impairments in DNA repair processes are highly associated with symptoms of aging. In fact, disorders that cause premature aging are typically caused by mutations in genes involved in the maintenance of our DNA. And at the cellular level, decreases in stem cells’ proliferative abilities, impairments in mitochondrial function, and proneness to protein misfolding can all contribute to aging. As scientists continue to detail these various processes, says Paul Robbins of the Scripps Research Institute, “the big question is, ‘At what step along all these pathways is the best place to intervene to try to promote healthy aging?’”
While diverse strategies—from caloric restriction to genetic manipulation—have ...












{kind=link}