
 DARRYL LEJA/NHGRI/SCIENCE SOURCE
DARRYL LEJA/NHGRI/SCIENCE SOURCE
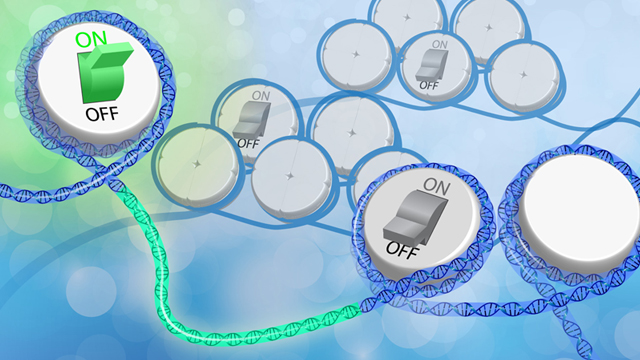
Chemical groups tacked onto DNA or histone proteins regulate how and when genes are expressed. Environmental signals can change the placement of these epigenetic tags, but researchers have had trouble pinning down how phenomena such as diet, inflammation, or social stress are converted into instructions that tweak gene functions.
Researchers have known for decades how some aspects of metabolism can wield epigenetic effects: breakdown products formed during sugar or protein digestion, for example, can be converted into chemical tags that epigenetically modify DNA or histones. But even a process as fundamental as turning glucose into cellular fuel can occur via distinct pathways that dynamically change based on a cell’s immediate environment and state. So a cancer cell and a healthy one might digest sugars in ...













